
Long-QT-Syndrom: „Mein Herz macht Extraschläge“
Im dritten Lebensjahr wurde beim heute 14-jährigen Severin* ein seltenes angeborenes Long-QT-Syndrom diagnostiziert. Seit der medikamentösen Einstellung auf Flecainid sind bei ihm keine schweren Herzrhythmusstörungen oder Ohnmachtsanfälle aufgetreten. Mit Serverins Diagnose konnten auch die Herzrhythmusstörungen seiner Mutter aufgeklärt werden, denen dieselbe genetische Variation zugrunde liegt.
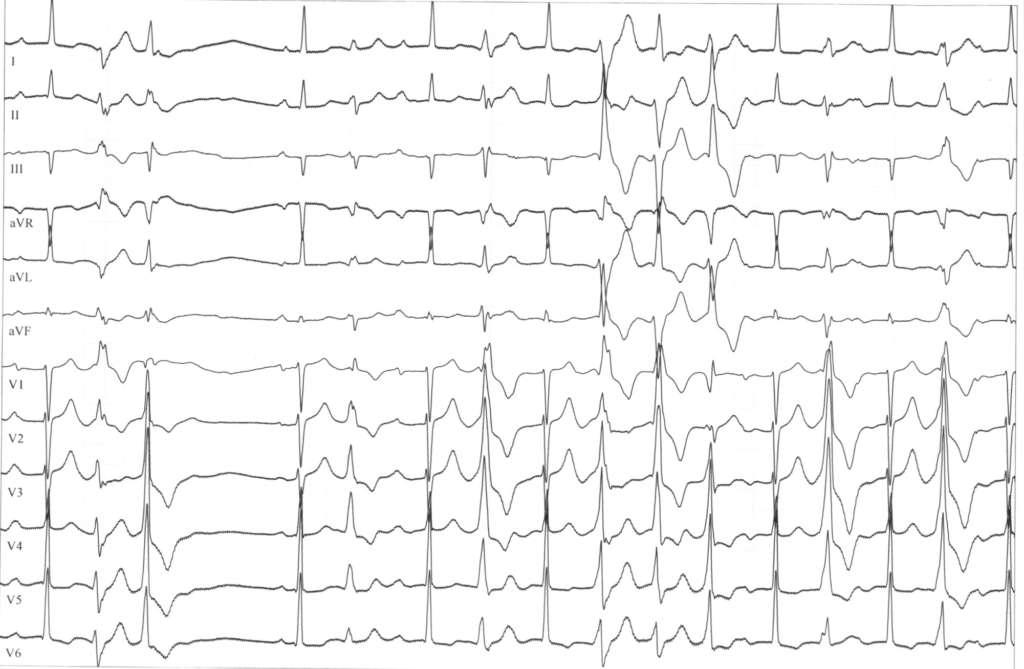
Severins EKG
Severin Rathners* erste Erinnerung an seine Erkrankung ist ein Ohnmachtsanfall im Kindergarten: „Ich bin einfach umgefallen, danach auf einem Stuhl aufgewacht und mit der Rettung in´s Krankenhaus gebracht worden“, erzählt er im gemeinsamen ZOOM-Gespräch. Der erste Verdacht in Richtung einer epileptischen Erkrankung bestätigte sich nicht, das auffällige EKG und später die genetische Untersuchung ergaben schließlich eine seltene Form einer angeborenen Ionenkanalerkrankung im Herz mit der Bezeichnung Long-QT-Syndrom Typ 7 oder Andersen-Tawil-Syndrom (siehe Fakten-Check).

OÄ Dr. Katrin Schamel
Mit der Einstellung auf Flecainid hat Severin seit nunmehr acht Jahren keine Ohnmachtsanfälle mehr erlitten. „Das Medikament kann die Extraschläge weitgehend unterdrücken: Bei Severins erstem Langzeit-EKG sahen wir 20 Prozent Extraschläge in 24 Stunden, heute hat er bei den Kontrollen 100 einzelne Extraschläge in 24 Stunden – das ist ein super Ergebnis.
Auch traten bei ihm nie wieder höhergradige Herzrhythmusstörungen auf“, berichtet Severins behandelnde Kinderkardiologin Dr. Katrin Schamel, die die Familie am Department für Kinderkardiologie an den Tirol Kliniken in Innsbruck betreut.
Severin ist sich heute darüber im Klaren, wie wichtig für ihn die dreimal tägliche Medikation ist. „Ansonsten hat sich bei mir nicht viel verändert“, meint der Teenager, der in seiner Freizeit am liebsten Sport ausübt und das ZOOM-Meeting früher verlässt, da er zum Training muss. „Meine Lehrer an der Schule, ebenso meine Mitschüler, Teamkollegen und Trainer sind darüber informiert, dass sie bei einem Ohnmachtsanfall sofort die Rettung rufen müssen, denn es könnte sich um eine Herzrhythmusstörung handeln“, sagt Severin. Zudem hat er sich einen speziellen Anhänger anfertigen lassen, in dem neben „Long QT“ auf Deutsch und Englisch ein entsprechender Notfall-Hinweis eingraviert ist. Er trägt diesen an einer Halskette immer dann, wenn er ohne seine Freunde unterwegs ist.
Zugleich mit Severins diagnostischer Abklärung wurde routinemäßig nach bekannten Herzerkrankungen in der Familie gefragt. „Tatsächlich hatte ich selbst im Kindesalter immer wieder Ohnmachtsanfälle und auch Herzrhythmusstörungen im Alter von 14 Jahren, die damals jedoch als eine harmlose Spinnerei des Herzens bezeichnet wurden“, erzählt Severins Mutter Judith Rathner*. Ein routinemäßiges EKG zum Zeitpunkt von Severins Diagnose ließ das diplomierte Pflegepersonal bei Herzspezialistin Schamel Alarm schlagen. Die genetische Untersuchung bestätigte daraufhin, dass Judith Rathner dieselbe genetische Variante wie Severin und damit Long-QT-Syndrom Typ 7 hat.
Gut informierte Patienten
Judith Rathner hatte zwischenzeitlich jedoch bereits einen Herzstillstand und musste reanimiert werden, sodass ihr ein Defibrillator implantiert wurde. Zweimal wurde sie seither korrekt geschockt, es wurden aber auch zwei sehr schmerzhafte Fehlschocks ausgelöst. Doch genau wie ihr Sohn betreibt die Literaturwissenschafterin in ihrer Freizeit gerne Sport und ist oft mit dem E-Bike oder auf Skiern unterwegs. „Moderater Freizeitsport ist bei Long-QT-Syndrom kein Problem“, bestätigt dazu Schamel.
Familie Rathner fühlt sich in allen medizinischen Fragen sehr gut unterstützt durch die Kinderkardiologin, die bei Severins Kontrollen stets ein offenes Ohr für alle Fragen hat. „Frau Schamel hat uns als Familie von Anfang an bestens betreut und uns viele Informationen gegeben“, berichtet Rathner. So weiß sie etwa auch, dass bestimmte Antibiotika die QTc-Zeit verlängern können und daher bei Patienten mit Long-QT-Syndrom vermieden werden sollten. „Zudem werde ich in meiner Apotheke diesbezüglich immer gut beraten und bin sehr für die Einführung von ELGA, die es möglich machen sollte, dass alle Fachärzte sofort darauf hingewiesen werden. Ich habe keinerlei Bedenken wegen dem Datenschutz und vor allem für Severin wäre es mir sehr wichtig.“
Schamel ergänzt: „Zum Glück sind es nur wenige Medikamentengruppen wie Makrolide, die eine relevante Verlängerung der QTc-Zeit bewirken, allerdings auch die meisten modernen Psychopharmaka – das kann für die Kollegen aus der Psychiatrie dann durchaus zur Herausforderung werden.“
Insgesamt unterstreicht die Kinderkardiologin die Bedeutung der Zusammenarbeit zwischen Kindern, Eltern und Ärzten: „Gerade bei so seltenen Erkrankungen sind Eltern immer sehr dankbar, wenn die Erkrankung einen Namen hat und eine Therapie zur Verfügung steht. Ganz wichtig ist es, dass die Medikamente regelmäßig eingenommen werden, denn sie können das Risiko für schwere Herzrhythmusstörungen verringern – wenn auch nicht zu 100 Prozent verhindern.“ Dies müsse gut erklärt werden, da die Erkrankung für die Patienten nicht sichtbar, sondern oft nur für Spezialisten am EKG zu erkennen ist.
*Namen geändert
Fakten-Check: Angeborene Long-QT-Syndrome
Als seltene, genetisch bedingte funktionelle Störungen verschiedener Ionenkanäle der Zellmembran des Herzens werden angeborene Long-QT-Syndrome beschrieben. Je nachdem, welche Ionenkanäle betroffen sind, werden Unterformen charakterisiert. Das Long-QT-Syndrom Typ 7 oder Andersen-Tawil-Syndrom wurde zunächst als wiederkehrende Muskelschwäche beschrieben, erklärt die Innsbrucker Kinderkardiologin Katrin Schamel. „Es geht zudem mit sehr heterogenen Phänotypen einher. Im Vergleich zu den klassischen Long-QT-Syndromen ist es eine Multisystemerkrankung mit periodischer Muskelschwäche und charakteristischen knöchernen Veränderungen wie z.B. spitz zulaufenden Fingern und einem zu kleinen Unterkiefer. Eine klinische Diagnose ist somit sehr schwierig.“ Dank der Fortschritte der Mutationsanalysen können die Syndrome heute jedoch gut abgegrenzt werden. Die Häufigkeit wird für das Long-QT-Syndrom Typ 7 mit mindestens 1:1.000.000 geschätzt, „die Dunkelziffer ist sicher höher“, meint Schamel. Die Vererbung erfolgt autosomal dominant, die Prävalenz aller Long-QT-Syndrome beträgt 1:2.000. Genaue Diagnostik und Therapie sind deshalb so wichtig, um medikamentös das Risiko für schwere Herzrhythmusstörungen zu reduzieren.