
Mikrovillöse Einschlusskrankheit (MVID): „Batuhan ist ein Überraschungskind“
Vor wenigen Wochen feierte Familie Avci Batuhans ersten Geburtstag. Aufgrund seiner seltenen mikrovillösen Einschlusskrankheit (MVID) kann der Vorarlberger Bub ausschließlich parenteral ernährt werden. Eine wichtige Stütze neben der medizinischen und sozialen Betreuung erfahren Batuhans Eltern durch den weltweiten Austausch mit anderen betroffenen Familien über soziale Medien.

Batuhans Start ins Leben war alles andere als einfach: Sofort nach der Geburt per vorzeitigem Kaiserschnitt musste er intensivmedizinisch behandelt werden: „Es hieß zunächst, seine Lunge ist krank, und er musste intubiert werden“, erzählt Batuhans Mutter Süreyya Avci. Nach einem Herzstillstand und weiteren Komplikationen durfte die Familie ihren Sohn erst am 20. Dezember 2021, fünfeinhalb Monate nach der Geburt, mit nach Hause nehmen. „Das war praktisch sein zweiter Geburtstag“, sagt Avci. Zu diesem Zeitpunkt stand Batuhans Diagnose bereits fest: Er leidet an der sehr seltenen Mikrovillösen Einschlusskrankheit MVID (siehe Fakten-Check) und kann keine Nahrung über den Darm aufnehmen.
„Da mein Mann und ich miteinander verwandt sind, wurde schon wenige Wochen nach der Geburt eine genetische Analyse gemacht. Das Team des Landeskrankenhauses Feldkirch hat gemeinsam mit dem Ärzteteam der Humangenetik Innsbruck nicht lockergelassen, bis sie die Diagnose hatten“, schildert Batuhans Mutter.
Humangenetische Sprechstunde
Die Vorarlberger Ärzte arbeiten unter anderem mit dem Zentrum für Humangenetik an der Medizinischen Universität Innsbruck zusammen. „Ich bin einmal pro Monat für ein paar Tage in Feldkirch und biete humangenetische Sprechstunden für Vorarlberg an“, sagt dazu die Fachärztin für Humangenetik Prof. Dr. Sabine Rudnik-Schöneborn. Bei einer dieser Gelegenheiten wurde ihr der damals erst neun Tage alte Batuhan vorgestellt. Es bestand der dringende Verdacht einer seltenen genetisch bedingten Erkrankung, auch weil er massive Magen-Darm-Probleme hatte und nichts zu sich nehmen konnte.

Prof. Dr. Sabine Rudnik-Schöneborn
„Da führte der Weg zur Diagnose nur über eine umfassende Exom-Analyse. Auch angesichts der Vorinformation einer Verwandtschaft zwischen den Eltern bestand die Hypothese einer autosomal rezessiv vererbten Erkrankung“, berichtet Rudnik-Schöneborn. Tatsächlich hat Batuhan von beiden Eltern dieselbe Mutation im MYO5B-Gen geerbt, sodass er kein funktionstüchtiges Gen trägt (homozygote Mutation). „Bei homozygoter MYO5B-Mutation wird der Bürstensaum der Darmschleimhaut nicht ausgebildet, sodass die Aufnahme der Nahrung nicht funktioniert. Von Geburt an können die erkrankten Kinder ohne eine permanente künstliche Ernährung nicht überleben, später kann eine Darm-Transplantation überlegt werden“, erläutert die Fachärztin.
Infusions-Management
„Die Diagnose war für uns ein Schock und hat unsere Welt komplett verändert“, setzt Batuhans Mutter Süreyya Avci fort. „Ich habe mich am Anfang gar nicht getraut, nach seiner Lebenserwartung zu fragen, aber dann habe ich mich im Internet und auf sozialen Medien auf die Suche gemacht. Es waren allerdings nur wenige und keine positiven Informationen, die ich gefunden habe, da es bei MVID bislang keine Heilung gibt.“ Über Instagram fand Familie Avci jedoch Kontakt zu zwei Familien in Dänemark und Deutschland, Erstere hat ihr Kind früh verloren, Zweitere schickte jedoch Bilder von einem großen Mädchen. „Solche Bilder geben natürlich Hoffnung, auch wenn ich mir dessen bewusst bin, dass mein Sohn schwer krank ist. Ich sage mir aber immer: Batuhan ist ein Überraschungskind. Wenn Probleme auftreten, dann packen wir zusammen und fahren ins Krankenhaus und schauen, wie es weitergeht. Wir versuchen jeden Tag so zu leben, als wäre es unser letzter, und genießen jede Sekunde miteinander.“
Vor allem zeigt sich Süreyya Avci voll des Lobes für die medizinische und soziale Betreuung in ihrer Heimatstadt Feldkirch, vor allem für das Team der Kinder-Intensivstation um Prim. Univ.-Prof. Dr. Burkhard Simma.
Der Umgang mit Infusionspumpe und Zentralkatheter ist für Familie Avci mittlerweile fixer Teil des Alltags, da Batuhan parenteral über einen zentralen Venenkatheter ernährt wird. Die aufwendige Einschulung der Familie war auch mit ein Grund für den langen Krankenhausaufenthalt. „Jeden Abend bereiten wir die Infusionslösung vor und hängen die Infusion an, wobei wir sehr sauber arbeiten müssen, um Batuhan vor Infektionen zu schützen.“ Den allergrößten Teil der Pflege übernimmt die Mutter, jedoch auch Batuhans vierjährige Schwester hilft schon mit: „Sie hat von Anfang an verstanden, dass ihr Bruder krank ist, und wenn er beim Anhängen der Infusion zappelt, darf sie seine Hände oder Füße halten“, berichtet Frau Avci.
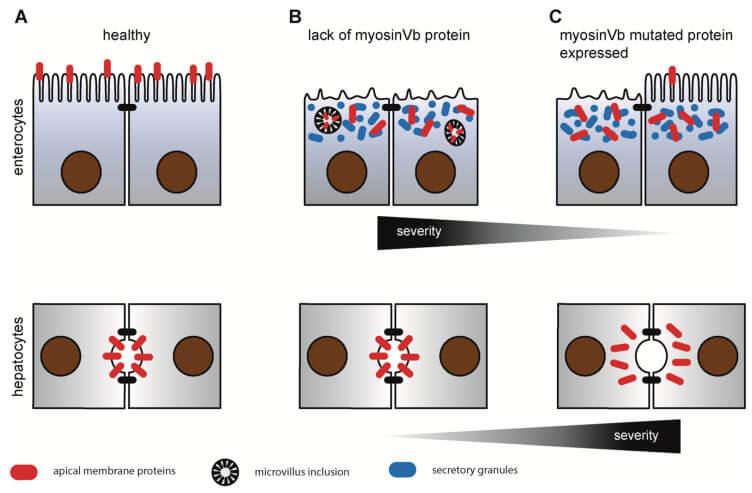
Abbildung: Schematischer Überblick zu den Veränderungen an Darm- und Leberzellen bei der MYO5B-bedingten MVID
A) Normalbefunde mit normaler epithelialer Differenzierung und regelrechter Lokalisation von apikalen Proteinen (rot) in den Bürstensaumzellen des Darms und den Gallenkanälchen in der Leber.
B) MYO5B-Mangel führt zu MVID ohne Leberbeteiligung. Abbau und Funktionsverlust des Bürstensaums, Ablagerung von sekretorischem Granulat (blau) und Einschlusskörperchen, fehlerhafte Lokalisation von apikalen Proteinen (rot), normale Leberfunktion.
C) Mutiertes MYO5B führt je nach Schweregrad der Mutation zu unterschiedlicher Darm- oder Leberfunktionsstörung. Fehlerhafte Lokalisation der apikalen Proteine (rot) in den Gallengängen verursacht eine primäre intrahepatische Cholestase.
Suche nach betroffenen Familien
Weiterführende medizinische Beratung bekommt die Familie mittlerweile auch am Zentrum für Chronisches Darmversagen und Intestinale Rehabilitation am Universitätsklinikum Tübingen, wo Batuhan bereits vorgestellt wurde. Am heimatlichen Krankenhaus in Feldkirch sind derzeit alle zwei Wochen Kontrollen nötig.
Während des gemeinsamen ZOOM-Gespräches ist Batuhan vom Nachmittagsschlaf aufgewacht und blickt mittlerweile am Arm seiner Mutter neugierig in die Kamera und hantiert mit seinem Spielzeug – es ist nicht zu übersehen, wie viel Freude Mutter und Sohn miteinander haben. „Klar kann ich nicht einfach mit ihm hinausgehen, und der Alltag ist sehr aufwendig. Ich bin jedoch froh, dass wir uns über das Internet mit anderen Familien mit MVID austauschen zu können“, sagt Avci.

In einer geschlossenen Facebook-Gruppe treffen sich mittlerweile rund 20 Familien aus aller Welt. Familie Avci habe sich entschlossen, mit diesem Bericht an die Öffentlichkeit zu gehen, auch da sie bislang in Österreich noch keine Kontakte zu anderen betroffenen Familien hat.
„Es gibt vermutlich weitere betroffene Familien, viele wollen aber nicht mit der Krankheit nach außen gehen, das ist nur verständlich“, sagt Fachärztin Rudnik-Schöneborn. „Wir müssen ehrlich sagen, dass die Sterberate bei dieser Erkrankung hoch ist. Allerdings lassen sich die Prognosen international schwer vergleichen, weil es nicht überall so rasch eine so gute kinderärztliche Versorgung wie hier in Österreich gibt.“ Zudem ist es erst in den letzten 10 bis 15 Jahren möglich geworden, die verschiedenen genetischen Ursachen von angeborenen Durchfallerkrankungen zu identifizieren und die klinischen Verläufe zu beschreiben. „Es gibt bislang keine Langzeiterfahrungen, aber wir lernen jeden Tag, mit diesen seltenen Diagnosen umzugehen.“
Humangenetische Beratung
Dringend nötig, so Rudnik-Schöneborn, sei ein Ausbau der humangenetischen Ambulanzen, die in Österreich bislang nur an universitären Zentren angesiedelt sind. Um der steigenden Nachfrage nach kompetenter genetischer Beratung besser gerecht zu werden, wurde in Innsbruck 2019 erstmals im deutschsprachigen Raum der Master-Studiengang „Genetisches und Genomisches Counselling“ für Nicht-Mediziner gestartet, dessen erste Absolventinnen nun in interdisziplinären Teams die Fachärzte für Medizinische Genetik unterstützen.
Erfreulich sind aus ihrer Sicht jedoch alle Initiativen, mit denen „die Stimme der Betroffenen mit jenen der Ärzte zusammengebracht werden“. Avci ergänzt: „Ich bin mir sicher, dass es in Zukunft möglich sein wird, Kindern wie Batuhan das Leben zu erleichtern. Vielleicht wird es zumindest einmal etwas geben, dass sie mit weniger Infusionen auskommen. Immer nur zu hören, dass es keine Heilung gibt, ist schwer, und doch versuchen wir den Moment gemeinsam zu genießen.“
Fakten-Check: Mikrovillöse Einschlusskrankheit (MVID)
Die angeborene MVID ist eine schwerwiegende Erkrankung des Verdauungstrakts, die dazu führt, dass Flüssigkeit und Nährstoffe nicht ausreichend über den Darm aufgenommen werden können. Ursache ist eine Entwicklungsstörung des Bürstensaums im Darm (Mikrovilli). Dünndarmbiopsien zeigen eine Zottenatrophie und abnormales Periodic acid Schiff (PAS)-Reaktion-positives Einschlussmaterial im Darmepithel, daher die Bezeichnung MVID. In den ersten Lebenswochen kommt es zu einer Gedeihstörung der betroffenen Kinder, die eine parenterale Ernährung unter Umgehung des Darmtrakts erforderlich macht.
Angeborene erblich bedingte Durchfallerkrankungen können durch verschiedene Gene verursacht werden. Das Myosin 5B (MYO5B)-Gen auf Chromosom 18 ist für die Organisation bzw. Ausrichtung der Bürstensaumzellen im Darm verantwortlich. In Abhängigkeit von der Schwere der verantwortlichen Erbgutveränderung können zahlreiche weitere Funktionsstörungen der inneren Organe auftreten. So kommt es bei der Hälfte der Patienten auch zu einer Leberbeteiligung aufgrund einer Fehlanlage der Gallengänge (intrahepatische Cholestase, siehe Abbildung). Eine ausführliche Beschreibung der vielfältigen Symptome wurde im letzten Jahr von einer internationalen Arbeitsgruppe unter Federführung der Universitäts-Kinderklinik Innsbruck veröffentlicht (Aldrian et al. J Clin Med 2021, J. Clin. Med. 2021, 10(3), 481).
MVID ist eine autosomal rezessive Erkrankung, bei der Symptome nur auftreten, wenn beide Genexemplare durch eine Mutation ihre Funktion einbüßen. Die Eltern tragen jeweils eine Mutation in einem Genexemplar und werden als heterozygote Anlageträger bezeichnet. Sie sind gesund, weil das zweite, normale Genexemplar für eine normale Funktion und Entwicklung des Verdauungstrakts ausreicht. Kinder von zwei heterozygoten Anlageträgern erkranken mit einer Wahrscheinlichkeit von 25 Prozent. Bei blutsverwandten Eltern steigt die Wahrscheinlichkeit für autosomal rezessive Krankheiten durch gemeinsame Vorfahren, die unerkannte heterozygote Anlageträger für entsprechende Mutationen sind.
Serie: Die Gesichter Seltener Erkrankungen
Seltene Erkrankungen frühzeitig zu erkennen und bestmöglich zu behandeln bzw. zu managen gehört zu den größten Herausforderungen der Medizin im dritten Jahrtausend. Mitunter sind es vielleicht nur zehn, zwölf Menschen in Österreich mit derselben Diagnose, die oft erst nach jahrelangen Wegen durch Ordinationen und Ambulanzen wissen, woran sie tatsächlich leiden. Die Diagnose erhielten sie meist von engagierten Ärztinnen und Ärzten, die auf den richtigen Pfad kamen und sich um Therapie und Management bemühen.
In der medonline-Serie in Kooperation mit dem Referat für Seltene Erkrankungen der Ärztekammer Wien wollen wir die Gesichter Seltener Erkrankungen vorstellen mit dem Ziel, das Bewusstsein dafür zu stärken: Seltene Erkrankungen sind zwar selten, aber es gibt sie! Mitunter sind sie aber viel zu wenig bekannt. Wir stellen Ihnen daher engagierte Ärztinnen und Ärzte und ihre Patientinnen und Patienten bzw. deren Eltern vor. Ihre Erfahrungen sollen dazu beitragen, Seltene Krankheiten besser bekannt zu machen und vielleicht rascher zur richtigen Diagnose und zur bestmöglichen Behandlung zu kommen.
Mag. Christina Lechner (Koordinierende Redakteurin) und Mag. Patricia Herzberger (Redakteurin medonline, CR Medical Tribune) mit Dr. Christoph Buchta (Ärztekammer Wien/Referat für Seltene Erkrankungen)
In Kooperation mit der Ärztekammer Wien
Referat für Seltene Erkrankungen